


scTHI is an R/Bioconductor package to identify active pairs of ligand-receptors from single cells in order to study,among others, tumor-host interactions. scTHI contains a set of signatures to classify cells from the tumor microenvironment.

Our software tool for Adaptive One-Class Gaussian Processes (OCGP) for oncology drug targets prioritization. See our paper for details.
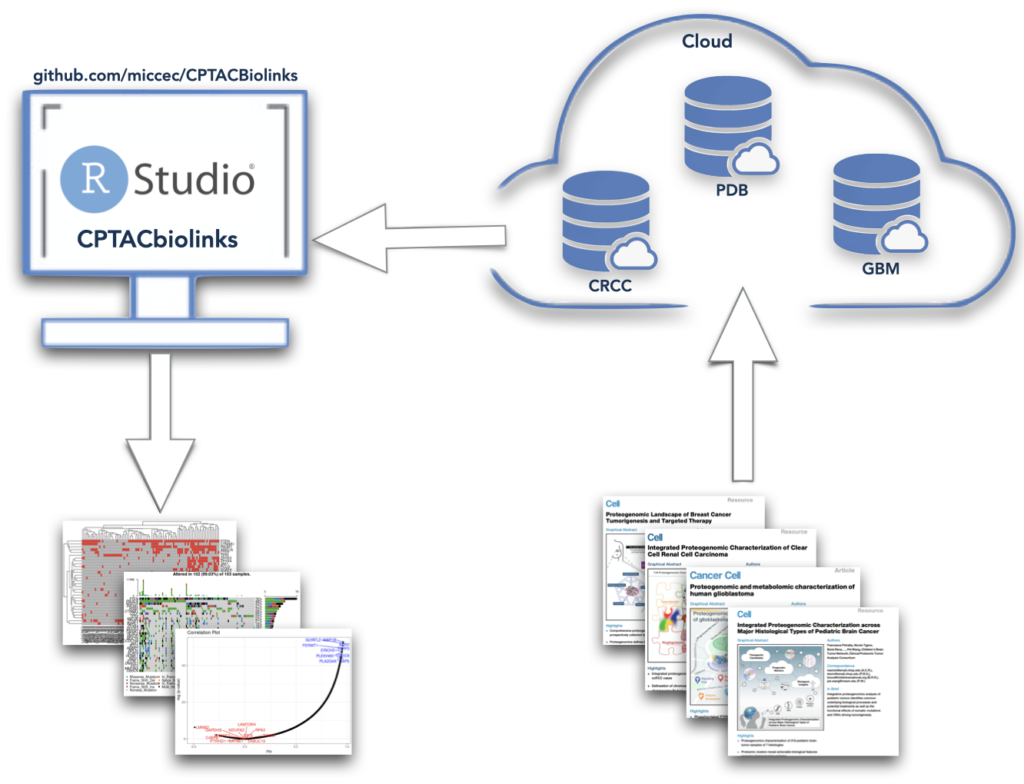
CPTACBiolinks is our tools to download and analyze CPTAC data.

RGBM: Regularized LS-TreeBoost & LAD-TreeBoost algorithm for Regulatory Network inference from any type of expression data (Microarray/RNA-seq etc). See our Mall et al. NAR paper.

ncrna-deep is a deep learning method to predict the function on short non coding RNA. The method is detailed in the paper “Deep learning predicts non-coding RNA functions from only raw sequence data“

TCGABiolinks is a pupular R/Bioconductor package for the integrative analysis of TCGA data developed with Antonio Colaprico, Thiago Silva and Houtan Noushmehr.

VEGA is a n R/Bioconductor package for Copy Number detection. It’s a segmentation algorithm based on the Mumford&Shah variational model.

ExomePipeline is our python notebook for running tumor-normal somatic variation calling for multiple samples.

TimeDelay-ARACNE is an R/Bioconductor package for gene regulatory network inference from time-course data based on Mutual Information.

GAIA is an R/Bioconductor package for the identification of recurrent Copy Number alteration from multiple tumors based on an homogeneous peel-off algorithm.

VEGAWES is the extension of VEGA segmentation algorithm for NGS data. See the our paper for details.

VegaMC is an R/Bioconductor package which extend the VEGA algorithm to multiple samples. Details are available in the Bioinformatics paper.